Was ist "Familiäre Hypercholesterinämie"?
Der Name „Familiäre Hypercholesterinämie“ setzt sich aus mehreren Bestandteilen zusammen: „Familiär“ heißt, dass die Störung vererblich ist, also von Eltern an ihre Kinder weitergegeben werden kann. „Hypercholesterinämie“ bedeutet so viel wie „zu viel Cholesterin im Blut“.
Die Familiäre Hypercholesterinämie (kurz: FH) ist also eine Erbkrankheit, die zu einer Störung des Fettstoffwechsels führt. Betroffene haben hohe oder sehr hohe Werte des schädlichen LDL-Cholesterins im Blut, was zu schweren Folgekrankheiten wie Schlaganfall oder Herzinfarkt führen kann.
Bei der Vererbung von Genen gilt vereinfacht gesagt: Für jedes Merkmal wie Haarfarbe, Schuhgröße oder auch die Höhe des natürlichen Cholesterinspiegels sind jeweils zwei Gene verantwortlich - ein Gen von der Mutter und ein Gen vom Vater. Die FH wird meist von einer Störung auf dem sogenannten LDLR-Gen verursacht, das wichtige Funktionen bei der Regelung des Cholesterinstoffwechsels erfüllt. Hat ein Kind von beiden Elternteilen jeweils ein defektes LDLR-Gen geerbt, so spricht man von der „homozygoten Form“ der FH. In beiden Genen besteht also der gleiche (griech. „homo“) Defekt. Diese Form ist sehr selten und tritt durchschnittlich bei einer Million Geburten nur einmal auf.
Die häufigere Form von FH ist die sogenannte „heterozygote Form“ – hier hat das Kind nur von einem Elternteil ein schadhaftes LDLR-Gen und vom anderen Elternteil ein gesundes Gen geerbt. Die beiden Gene sind also unterschiedlich (griech. „hetero“). Von dieser Form der FH ist in Deutschland etwa eine von 500 Personen betroffen. Die Familiäre Hypercholesterinämie gehört in den Industrieländern damit zu den am weitesten verbreiteten genetischen Störungen.
Cholesterin – lebenswichtig und doch gefährlich
Ganz ohne Cholesterin könnte unser Körper nicht überleben. Wir benötigen das Blutfett für eine Vielzahl von Prozessen in unserem Organismus. So ist Cholesterin ein wichtiger Baustein für Zellmembranen und das Ausgangsprodukt für verschiedene Hormone und Gallensäuren. Gleichzeitig ist zu viel Cholesterin aber äußerst gefährlich für unseren Körper – wie kann das sein?
Hier ist zunächst etwas Grundlagenwissen nötig: Jede menschliche Zelle benötigt Cholesterin. Um an das Cholesterin zu gelangen, besitzen gesunde Zellen Rezeptoren, mit denen sie das Cholesterin aus dem Blutkreislauf herausfiltern können. Allerdings lässt sich Cholesterin nicht einfach im Blut transportieren, denn in seiner Reinform ist es eine fettähnliche Substanz, die kaum wasserlöslich ist. Daher wird es für den Transport im Körper an bestimmte Trägereiweiße (lat. Proteine) gebunden. Solche Verbindungen aus Cholesterin und Proteinen heißen Lipoproteine.
Mit Hilfe dieser Lipoproteine lässt sich das Cholesterin also im Blutkreislauf transportieren. Dabei kommen verschiedene Lipoproteine zum Einsatz, die nach ihrer Dichte unterschieden werden. Zu den wichtigstens Lipoproteinen gehören das bereits erwähnte LDL (Low Density Lipoprotein, zu Deutsch: Lipoprotein niedriger Dichte) und das sogenannte HDL (High Density Lipoprotein, zu Deutsch: Lipoprotein hoher Dichte). Beide Lipoproteine übernehmen wichtige Rollen beim Transport des Cholesterins – das LDL transportiert Cholesterin zu den Zellen hin, das HDL transportiert überschüssiges Cholesterin dagegen zurück zur Leber, wo es aus dem Blutkreislauf entfernt wird. In der Leber sitzen deshalb normalerweise mit Abstand am meisten LDL-Rezeptoren.
Benötigt eine Zelle Cholesterin, aktiviert sie im Normalfall ihre LDL-Rezeptoren, die das LDL-Cholesterin dann aus dem Blutkreislauf herausfiltern. Bei Menschen mit Familiärer Hypercholesterinämie ist dieser Mechanismus aber gestört – sie können aufgrund ihres genetischen Defekts nicht genug LDL-Rezeptoren bilden und das LDL-Cholesterin reichert sich daher im Blut an.
Aus dem Blutkreislauf gelangt das LDL auch in die Zellwände der Arterien – und ab hier wird es problematisch. Körpereigene Freßzellen reagieren auf das LDL-Cholesterin und versuchen, es zu beseitigen. Dabei überladen sich die Freßzellen aber mit dem gespeicherten Fett und werden zu sogenannten Schaumzellen. Diese Schaumzellen wiederum gehören zu den Hauptauslösern für Arteriosklerose, umgangssprachlich auch Arterienverkalkung genannt.
Bei Menschen mit Familiärer Hypercholesterinämie beginnt die Arteriosklerose oft schon in jungen Jahren – das heimtückische daran ist, dass dieser Prozess meist völlig schmerzfrei und ohne Symptome verläuft. Vielfach bemerken Patienten erst etwas von der FH, wenn es zu einem schwerwiegenden Ereignis kommt, etwa einem Herzinfarkt oder einem Schlaganfall. Bei Betroffenen mit der homozygoten Form kann das schon im Teenager-Alter geschehen! Deshalb setzt sich CholCo für ein regelhaftes Screening der Lipidwerte bei Kindern ein.
1. Symptome und Diagnose
Das heimtückische an der Familiären Hypercholesterinämie ist, dass viele Betroffene zunächst nichts von ihrer Erkrankung bemerken – zu hohe Blutfettwerte verursachen keine Beschwerden. Und nur bei wenigen Patienten sind äußerliche Anzeichen zu bemerken. Vor allem junge Patienten haben oft noch keine großen Beschwerden, obwohl ihre Blutgefäße durch den hohen Cholesterinspiegel schon großen Belastungen ausgesetzt sind.
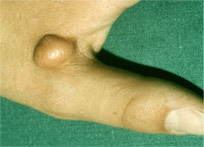 Bei einigen Betroffenen zeigt sich die FH in Form von sichtbaren Merkmalen am Körper. Dazu zählen in erster Linie Hautveränderungen, sogenannte Xanthome. Diese meist gelben und unregelmäßigen Knötchen treten bevorzugt in Achilles- und Fingerstrecksehnen, an den Ellenbogen und an den Kniescheiben auf. Auf der linken Seite sehen Sie ein typisches Xanthom an der Hand.
Bei einigen Betroffenen zeigt sich die FH in Form von sichtbaren Merkmalen am Körper. Dazu zählen in erster Linie Hautveränderungen, sogenannte Xanthome. Diese meist gelben und unregelmäßigen Knötchen treten bevorzugt in Achilles- und Fingerstrecksehnen, an den Ellenbogen und an den Kniescheiben auf. Auf der linken Seite sehen Sie ein typisches Xanthom an der Hand.
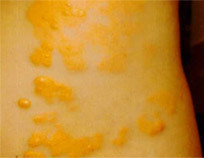 Auf diesem Bild ist ein Xanthom abgebildet, das sich an der Hüfte eines fünfjährigen Kindes gebildet hat. In seltenen Fällen treten bei FH auch Veränderungen in den Augen auf, in Form einer gelb-weißen, ringförmigen Trübung am Rand der Hornhaut. Meist bleibt aber eine FH unsichtbar und wird nur aufgrund klinischer Parameter, also beispielsweise aufgrund der Blutwerte, diagnostiziert.
Auf diesem Bild ist ein Xanthom abgebildet, das sich an der Hüfte eines fünfjährigen Kindes gebildet hat. In seltenen Fällen treten bei FH auch Veränderungen in den Augen auf, in Form einer gelb-weißen, ringförmigen Trübung am Rand der Hornhaut. Meist bleibt aber eine FH unsichtbar und wird nur aufgrund klinischer Parameter, also beispielsweise aufgrund der Blutwerte, diagnostiziert.
Wie wird FH diagnostiziert?
Die klinische Diagnose der FH basiert auf der Erhebung der Krankheitsgeschichte des Betroffenen und seiner Familie (Anamnese), einer körperlichen und genetischen Untersuchung und der Bestimmung der Blutfettwerte. Die Blutfettwerte können in unterschiedlichen Einheiten angegeben werden, entweder in Milligramm pro Deziliter (mg/dl) oder in Millimol pro Liter (mmol/l), die Aussagekraft der Werte ist aber identisch.
Wichtig für die Diagnose ist die Unterscheidung zwischen den verschiedenen Arten des Cholesterins, einmal dem sogenannten „High Density Lipoprotein-Cholesterin“ oder kurz HDL-Cholesterin und zum anderen dem „Low Density Lipoprotein-Cholesterin“ oder LDL-Cholesterin.
Das LDL wird bei Routineuntersuchungen dabei meist nicht direkt gemessen, sondern nach der sogenannten Friedewald-Formel errechnet, da die direkte Messung vergleichsweise aufwendig ist. Deshalb werden zunächst die einfacher zu bestimmenden Werte für das Gesamtcholesterin, das HDL-Cholesterin und die sogenannten Triglyceride erhoben und anschließend auf Basis dieser Formel verrechnet:
LDL-Cholesterin = Gesamtcholesterin - HDL-Cholesterin - Triglyceridwert/5
Diese Formel gilt nur, wenn die Triglyzeride unter 400 mg/dl liegen.
Bei der heterozygoten Form können die Cholesterinwerte zwischen 270-550 mg/dl liegen, bei der homozygoten Form sogar zwischen 650-1000 mg/dl.
Wann liegt eine FH vor?
Nach internationalen Kriterien ist definitiv vom Vorliegen einer Familiären Hypercholesterinämie auszugehen, wenn bei:
Kindern unter 16 Jahren das Gesamt-Cholesterin größer als 270 mg/dl (6,7 mmol/l) ist oder das LDL-Cholesterin größer als 160 mg/dl (4,0 mmol/l) ist.
Erwachsenen das Gesamt-Cholesterin größer als 300 mg/dl bzw. 7,5 mmol/l ist oder das LDL-Cholesterin mehr als 200 mg/dl bzw. 5 mmol/l beträgt und zusätzlich Sehnen-Xanthome bei dem Patienten oder Verwandten ersten oder zweiten Grades auftreten oder der DNA-basierte Nachweis einer genetischen Mutation erbracht wurde.
Eine mögliche FH ist definiert bei:
Kindern unter 16 Jahren, wenn das Gesamt-Cholesterin größer als 270 mg/dl bzw. 6,7 mmol/l ist oder das low-density-Lipoprotein-Cholesterin (LDL-C) größer als 160 mg/dl bzw. 4,0 mmol/l ist.
Erwachsenen, wenn das Gesamt-Cholesterin größer als 300 mg/dl bzw. 7,5 mmol/l ist oder das LDL-C mehr als 200mg/dl bzw. 5 mmol/l beträgt und beim Vorliegen von mindestens einem der folgenden Punkte: Eine Familienanamnese für Herzinfarkt, also wenn bei einem Verwandten zweiten Grades unter 50 Jahren oder bei einem Verwandten ersten Grades unter 60 Jahren schon einmal ein Herzinfarkt aufgetreten ist.
Wann soll man an FH denken?
Der Verdacht auf eine Familiäre Hypercholesterinämie ergibt sich meist aus einem der folgenden Anlässe:
- Wenn bei einer Routineuntersuchung ein viel zu hoher Wert für das LDL-Cholesterin festgestellt wurde oder
- Wenn man in vergleichsweise jungem Alter einen Herzinfarkt oder Schlaganfall erleidet oder
- Wenn ein Familienscreening (Familienuntersuchung) durchgeführt wird und ein erhöhter LDL-Cholesterinspiegel auffällt.
Leider tritt der erste Fall, also die Entdeckung nach einer Routineuntersuchung, viel zu selten auf, da nur bei einem kleinen Teil der Menschen der LDL-Cholesterinspiegel ohne speziellen Anlass getestet wird. Weil die Familiäre Hypercholesterinämie aber vergleichsweise häufig ist, fordert CholCo eine flächendeckende Bestimmung des LDL-Cholesterins schon bei Kindern, am besten in Form eines regelhaften Screenings (Untersuchung Gesunder). Eine Bestimmung der benötigten Blutwerte ist weder teuer noch aufwendig, pro Test sind nur einige Euro nötig. Der mögliche Nutzen ist dagegen enorm: Wenn vermieden werden kann, dass Patienten mit Familiärer Hypercholesterinämie ihre Diagnose zu spät erhalten, ließen sich viele Folgeschäden verhindern. Ziel muss es sein, die FH vor Entstehung der Folgekrankheiten zu diagnostizieren und zu behandeln!
Der zweite genannte Anlass, die Diagnose nach einem schwerwiegenden Ereignis wie Herzinfarkt oder Schlaganfall, ist sicherlich der häufigste. Obwohl dann schon eine schwere, lebensgefährliche Krankheit vorliegt, ist es noch nicht zu spät, sich auf Familiäre Hypercholesterinämie untersuchen zu lassen. Schließlich muss nach einem Herzinfarkt oder Schlaganfall mit allen Kräften versucht werden, das Fortschreiten der Arteriosklerose zu verhindern; das LDL muss rasch und zielwertgerecht gesenkt werden! Weitere Informationen zu Zielwerten finden Sie auch im folgenden Punkt "Behandlung und Therapie".
Der dritte Anlass, der Sie und Ihren Arzt für die Familiäre Hypercholesterinämie sensibilisieren sollte, ist die Erkrankung von Blutsverwandten – hier sollten unbedingt die nächsten Verwandten auf eine mögliche Erkrankung hin untersucht werden.
2. Behandlung und Therapie
Da es sich bei der Familiären Hypercholesterinämie um eine Erbkrankheit handelt, lässt sie sich nicht heilen. Dank moderner Medikamente und Behandlungsmöglichkeiten ist die Krankheit aber zumindest in den meisten Fällen beherrschbar, so dass Betroffene ein weitgehend normales Leben führen können. Entscheidend für eine erfolgreiche Therapie ist es, die FH so früh wie möglich zu erkennen – so können schwere Gefäßschäden (Arteriosklerose) verhindert werden. Wer die Diagnose „Familiäre Hypercholesterinämie“ erhält, sollte daher auch unbedingt nahe Blutsverwandte auf die Krankheit aufmerksam machen, um eine mögliche Erkrankung abzuklären. Es kann hilfreich sein, einen Stammbaum der Familie zu zeichnen, um die Vererbungswege zu erkennen.
Wesentlich für die Therapie einer Familiären Hypercholesterinämie ist die regelmäßige Kontrolle der Blutfettwerte und die damit verbundene Bemühung, die Cholesterinwerte innerhalb der Normwerte zu halten. Dazu bieten sich mehrere Möglichkeiten. Zu den wichtigsten gehören die konsequente Befolgung von Ernährungsrichtlinien, sportliche Betätigung, die Einnahme cholesterinsenkender Medikamente und die Lipid-Apherese. Als anzustrebende Zielwerte gelten:
- Gesamtcholesterin weniger als 200 mg/dl
- LDL-Cholesterin weniger als 130mg/dl
- HDL-Cholesterin mehr als 40 mg/dl bei Männern und mehr als 45 mg/dl bei Frauen.
Wichtig: Diese Zielwerte sind Richtwerte, die gegebenenfalls je nach Vor- oder Begleiterkrankung vom behandelnden Arzt individuell festgelegt werden.
Therapeutische Möglichkeiten
An dieser Stelle finden Sie einen kurzen Überblick über die therapeutischen Möglichkeiten – für detaillierte Infos folgen Sie einfach den Links (derzeit noch nicht aktiviert).
Zwei Bausteine, die auf jeden Fall zur Therapie gehören sollten, sind die richtige Ernährung und ausreichend Sport. In den meisten Fällen der Familiären Hypercholesterinämie wird auch eine medikamentöse Therapie nötig sein. Hier gilt es, das richtige Medikament und die richtige Dosierung zu finden, mit der Ihr Cholesterinspiegel auf den gewünschten Zielwert gebracht werden kann. Bleiben die Werte trotz dieser Maßnahmen zu hoch, besteht akute Gefahr für die Gesundheit – bis vor wenigen Jahrzehnten konnte nichts für derartig betroffene Patienten unternommen werden. Seit Mitte der 1980er-Jahre steht aber mit der sogenannten Lipid-Apherese eine neue Möglichkeit zur Verfügung, mit der auch schwere Fettstoffwechselstörungen behandelt werden können. Die Apherese ist eine Art Blutwäsche, ähnlich einer Dialyse, bei der die schädlichen Blutfettbestandteile maschinell aus dem Körper entfernt werden können. Weitere Informationen dazu finden Sie im entsprechenden Unterkapitel. Dort finden Sie ebenfalls einen Link auf hilfreiche Adressen, wo Sie noch weitere Informationen zur Familiären Hypercholesterinämie und anderen Stoffwechselerkrankungen finden.
Grundsätzlich gilt: Die Art der Therapie muss immer individuell abgestimmt werden, denn jede Erkrankung ist anders und Betroffene reagieren unterschiedlich auf Nahrungsmittel oder Medikamente. Scheuen Sie sich nicht, mit Ihrem Arzt über Ihre persönlichen Therapiemöglichkeiten zu sprechen!
- Ernährung »
- Sport »
- Medikamente »
- Lipid-Apherese »
- Behandlung vom Fachmann »
- Wichtige Adressen und Ansprechpartner »
3. Folgeerkrankungen
Ein chronisch erhöhter Cholesterinwert bereitet zunächst keine körperlichen Beschwerden. Gefährlich wird die FH erst durch die schwerwiegenden Folgekrankheiten, die mit dem dauerhaft hohen LDL-Cholesterin-Spiegel einhergehen.
Besonders die Blutgefäße sind gefährdet: Die erhöhten LDL-Cholesterin-Werte begünstigen Erkrankungen die Arteriosklerose, im Volksmund auch „Arterienverkalkung“ genannt. Hier entstehen im Laufe der Zeit Ablagerungen in den empfindlichen Gefäßwänden, die das Gefäß verengen und den Blutfluss massiv einschränken können. An derart verengten Stellen bleiben auch Blutgerinnsel viel schneller hängen und diese Ablagerungen können aufplatzen – die Folge ist ein akuter Gefäßverschluss, der zum Herzinfarkt oder Schlaganfall führen kann.
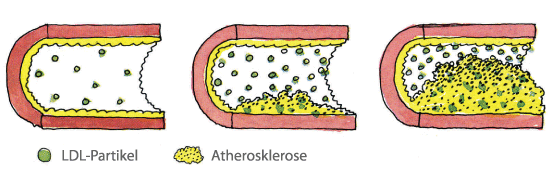
Patienten mit FH (defekte Zelle) haben weniger LDL-Rezeptoren, die LDL-Cholesterin
aus dem Blut entfernen können.
Bei der FH sind die Cholesterinwerte von Geburt an stark erhöht, deshalb sind die Blutgefäße schon früh einer hohen Belastung ausgesetzt - teilweise erleiden schon kleine Kinder oder Teenager den ersten Herzinfarkt oder den ersten Schlaganfall. Darum setzt sich CholCo für ein Screening bei Kindern ein, um das Erkrankungsrisiko so früh wie möglich zu erkennen.
4. Schwangerschaft und Stillzeit
Patienten mit Familiärer Hypercholesterinämie haben aufgrund ihres gestörten Cholesterinhaushaltes ein sehr hohes Risiko, an Herz- und Kreislaufleiden zu erkranken. Zur langfristigen Senkung des Cholesterinspiegels reichen eine gesunde Ernährung und Sport alleine häufig nicht aus, deshalb wird bei vielen Patienten eine zusätzliche Behandlung mit Medikamenten nötig, selbst wenn noch keine Anzeichen für Herz- und Gefäßschäden bestehen.
Rechnerisch liegt in Deutschland bei etwa 16 000 Frauen zwischen 20 und 40 Jahren eine Familiäre Hypercholesterinämie vor - davon theoretisch bei ein oder zwei Frauen in homozygoter Merkmalsform, also mit defekten Genen von beiden Elternteilen. Durch hormonelle Veränderungen kann der Cholesterinwert in der Schwangerschaft noch einmal auf das Doppelte ansteigen, die Triglyzeride können sogar dreimal so hoch wie vor der Schwangerschaft werden. Betroffene Frauen fragen sich dann natürlich, ob die Pharmakotherapie, also die Behandlung mit cholesterinsenkenden Medikamenten, auch während der Schwangerschaft und in der Stillzeit sicher ist?
Noch keine gesicherten Erkenntnisse
Zur medikamentösen Behandlung der Hypercholesterinämie stehen Medikamente zur Verfügung, die im Körper chemisch Einfluss auf den Stoffwechsel nehmen und Substanzen, die bereits im Darm wirken, ohne in den Blutkreislauf aufgenommen zu werden (siehe Kapitel „Medikamente“). Prinzipiell sind nur Auswirkungen von Medikamenten ein mögliches Problem, die in den Blutkreislauf aufgenommen werden, also von sogenannten resorbierbaren Medikamenten.
Zu Verträglichkeit und Sicherheit dieser resorbierbaren Cholesterinsenker kurz vor und während der Schwangerschaft liegen zwar Einzelbeobachtungen vor. Diese erlauben allerdings noch kein wissenschaftlich fundiertes Urteil. Einige tierexperimentelle Studien sprechen jedoch für die Möglichkeit von Schäden für das Kind. Exakte Studiendaten zur Behandlung mit Cholesterinsenkern in der Schwangerschaft fehlen aber leider - das bringt den beratenden Arzt in ein Dilemma: Welches Risiko besteht für das Kind? Ist eine während der Schwangerschaft unbehandelte Hypercholesterinämie ein Problem für die Mutter oder sogar für das Kind?
Um das Risiko für Mutter und Kind zu minimieren, gilt daher momentan folgende Empfehlung:
Resorbierbare Medikamente wie Statine, Ezetimib, Fibrate und Nikotinsäure gelten in Schwangerschaft und Stillzeit als nicht anwendbar (also als kontraindiziert) und werden nicht empfohlen.
So ergibt sich nach derzeitigem Wissensstand die Empfehlung, mindestens vier Wochen oder eher sogar drei Monate vor der Empfängnis sowie in der Schwangerschaft und der Stillzeit Medikamente aus den genannten Substanzgruppen auszusetzen. Als möglich gilt die Einnahme von nicht vom Körper aufgenommenen Stoffen, sogenannten Anionenaustauschern.
Es ist nicht sicher, welche Bedeutung eine ausgeprägte Hypercholesterinämie in der Schwangerschaft für Mutter oder Kind hat. Auf alle Fälle wird der Einsatz der LDL-Apherese bei Vorliegen der Stoffwechselstörung in homozygoter Merkmalsform diskutiert. Die Entscheidung hängt von der Schwere des klinischen Bildes ab. Einzelbeobachtungen wecken die Sorge, dass sich für das werdende Kind ein höheres Gefäßrisiko ergeben könnte, wenn es bereits während der Schwangerschaft einem übermäßig hohen Cholesterinspiegel ausgesetzt ist.
Es gibt auch Erfahrungsberichte von nicht geplanten Schwangerschaften, bei denen Mütter während des Beginns der Schwangerschaft regelmäßig Statine zu sich nahmen. Schadenshinweise waren dabei aber so minimal, dass betroffener Mütter in diesem Fall meist beruhigt werden können.
Wichtig: Diese Empfehlungen ersetzen auf keinen Fall einen Gang zum Arzt! Sollten Sie planen, schwanger zu werden und an Familiärer Hypercholesterinämie leiden oder sind Sie bereits schwanger, sollten Sie unbedingt einen Arzt hinzuziehen – idealerweise einen Lipidspezialisten.
5. FH bei Kindern
Eine Familiäre Hypercholesterinämie lässt sich gleich nach der Geburt diagnostizieren - bereits aus dem Nabelschnurblut kann die Diagnose gestellt werden. Oft ist die Diagnose in der Praxis ein Zufallsbefund im Rahmen anderer Untersuchungen oder Teil einer Familienuntersuchung nach Diagnose einer FH bei nahen Angehörigen. In seltenen Fällen wird die Familiäre Hypercholesterinämie auch bei der Feststellung von Xanthomen diagnostiziert.
 Zum unselektiven Screening auf das Vorliegen einer FH, also der automatischen Routineuntersuchung von Cholesterin im Rahmen einer Vorsorgediagnostik, gibt es unterschiedliche Auffassungen. Einerseits lässt sich aus wissenschaftlichen Studien noch nicht ableiten, ab wann bei Kindern Medikamente zur Cholesterinsenkung eingesetzt werden sollten und dürfen. Andererseits zeigt die Erfahrung, dass die Folgen der FH wesentlich ausgeprägter sind, wenn die Krankheit lange Zeit unerkannt bleibt.
Zum unselektiven Screening auf das Vorliegen einer FH, also der automatischen Routineuntersuchung von Cholesterin im Rahmen einer Vorsorgediagnostik, gibt es unterschiedliche Auffassungen. Einerseits lässt sich aus wissenschaftlichen Studien noch nicht ableiten, ab wann bei Kindern Medikamente zur Cholesterinsenkung eingesetzt werden sollten und dürfen. Andererseits zeigt die Erfahrung, dass die Folgen der FH wesentlich ausgeprägter sind, wenn die Krankheit lange Zeit unerkannt bleibt.
Nach Empfehlungen der Arbeitsgemeinschaft für Pädiatrische Stoffwechselstörungen (APS) in der Deutschen Gesellschaft für Kinderheilkunde und Jugendmedizin kommt die Cholesterinbestimmung ab dem 3. Lebensjahr in Frage, wenn bei Angehörigen Hinweise für eine vorzeitige koronare Herzkrankheit (KHK) und/oder erhebliche Fettstoffwechselstörungen vorliegen – vorzeitig bedeutet, dass Betroffene jünger als 60 Jahre waren.
Für ein generelles Cholesterinscreening wird die Cholesterinbestimmung anlässlich der letzten bislang von der Krankenkasse bezahlten pädiatrischen Vorsorgediagnostik U9 (ab 5 Jahren) angeregt. Die aktuellste Stellungnahme liegt von der amerikanischen National Lipid Association vor. Bei einer entsprechenden Familienvorgeschichte soll demnach bereits ab dem 2. Lebensjahr auf das Vorliegen der Stoffwechselstörung untersucht werden. Für ein universelles Screening sollten Kinder zwischen 9 und 11 Jahre alt sein, weil meistens erst dann der früheste Zeitpunkt für eine medikamentöse Behandlung gekommen ist.
Besonderheiten der Therapie von Kindern
Grundsätzlich ist es sehr wichtig, dass die Kinder von frühester Kindheit an einen gesunden Lebensstil lernen und diesen ganz einfach als normalen Lebensstil kennen. Das umfasst die ausgewogene Ernährung, viel Bewegung und lebenslangen Nikotinverzicht. Hier ist das Vorbild der Eltern ganz entscheidend.
Unter Fachleuten besteht weitgehend Konsens zur medikamentösen Therapie bei Kindern und Jugendlichen, obwohl diese noch nicht durch längere Studien bestätigt wurde. Häufig zeigen sich schon im Kindesalter die Auswirkung einer Familiären Hypercholesterinämie mit Verdickungen der Wände der Halsschlagadern, die mit Ultraschall erfassbar sind. Nach der Einnahme von Statinen nehmen diese Verdickungen oft ab, was zumindest als indirekter Hinweis auf den Nutzen gedeutet wird. Weitere Erfahrungen internationaler Ärzte und Forschergruppen zeigen ebenfalls positive Wirkungen der medikamentösen Therapie im Kindesalter. Die Behandlung sollte ab 8. Lebensjahr in Betracht gezogen werden und eine 50%ige LDL- Cholesterinsenkung erreichen. Wenn ein Kind sehr schwer betroffen ist, sollte die Therapie auch schon früher beginnen.
Medikamentös werden schon seit den 60er Jahren sogenannte Anionenaustauscher eingesetzt. Als günstig gilt, dass diese Substanzgruppe nicht in den Stoffwechsel eingreift und damit im Wachstumsalter nicht zu Veränderungen führt. Als Nachteil wird von einigen Patienten empfunden, dass relativ große Mengen von diesen Medikamenten genommen werden müssen.
Am meisten durchgesetzt hat sich die Therapie mit der Substanzgruppe der Statine, die die Synthese (Bildung) von Cholesterin vor allem in den Leberzellen hemmen. Die Einnahme ist einfach und der Effekt ist meist stärker als bei den Anionenaustauschern. Die Wirkung der Statine muss häufig durch Einsatz eines Cholesterinresorptionshemmers verstärkt werden.
Bei sehr schweren Verläufen der FH kommt eine Art „Blutwäsche“ in Frage, die sogenannte
Lipid-Apherese, die wöchentlich zusammen mit einer medikamentösen Kombinationstherapie eingesetzt wird. In schwersten Fällen sind auch Lebertransplantationen zur Therapie der Familiären Hypercholesterinämie vorgenommen worden, weil dadurch dem Organismus wieder funktionsfähige Rezeptoren für den LDL-Abbau übertragen werden können und so Herzinfarkte im Kindesalter verhindert werden können – dies betrifft aber nur Fälle mit homozygoter Ausprägungsform und ist auch hier extrem selten, da die Lipid-Apherese sehr effektiv und weniger einschneidend ist.

